Chorea Huntington
Synonyymit laajemmassa merkityksessä
- Vitus-tanssi (vulg.)
- Huntingtonin tauti
Englanti: Huntingtonin tauti, suuri korea.
Määritelmä
Perinnöllinen sairausjohtaa tuhoamiseen Aivosolut tietyillä aivoalueilla tajuttomien liikkumistaitojen pitämistä ja tukemista. Tauti esiintyy yleensä ikäryhmissä 35-50. Elämävuosi ja ilmaistaan
- Liikuntahäiriöt, kuten raajojen tahattomat, salamannopeat, liukuvat liikkeet
- irvistäen
- Älyllisen toimintakyvyn ja
- Henkilöllisyyden väheneminen.
Mikä on Huntingtonin taudin elinajanodote?
Normaaliin väestöön verrattuna Huntingtonin tautia sairastavien potilaiden elinajanodote on lyhyempi. Kuinka korkea yleinen elinajanodote on, vaihtelee suuresti henkilöittäin. Tämä riippuu toisaalta puhkeamisen iästä ja toisaalta taudin kulusta. Yleensä ensimmäiset oireet ilmenevät 30–40-vuotiaina. Lähes puolet kärsivistä kuolee taudin kymmenen ensimmäisen vuoden aikana. 15. sairausvuoden jälkeen vain 25% on edelleen elossa. Kymmenessä prosentilla tapauksista sairaus kesti myös yli 20 vuotta. Periaatteessa naisilla on keskimäärin hieman pidempi sairauden kesto kuin miehillä. Mitä aikaisemmin tauti esiintyy, sitä vakavampi kulku on. Huntingtonin tautia sairastavien potilaiden elinajanodote on keskimäärin 40-50 vuotta, vaikka taudin myöhäisessä alkamisessa ikä olisi noin 60 vuotta.
epidemiologia:
Huntingtonin taudin esiintymistiheys on 5-10 - 100 000, perintö on autosomaalisesti hallitseva. Tämä tarkoittaa, että sairastuneiden lapsilla on 50% riski sairastua itse tautiin.
oireet:
Vaikuttajilla on lihaksen heikkous, samalla laaja, salaman kaltaiset liukumisliikkeet raajoista, jotka pahenevat emotionaalisen jännityksen ollessa ja jotka ilmaantuvat harvoin unen aikana.
Syynä tähän on se, että tarvittavat impulssit eivät pysty estämään liikettä. Liikkeiden häiriintynyt koordinaatio ilmenee edelleen grimasoinnissa, nielemishäiriöissä ja puhevaikeuksissa. Chorea Huntington etenee, kun potilaalla etenee, sillä on vaikea kävellä, koordinoida silmien liikkeitä ja tulla kyvyttömäksi pitämään ulosteita ja virtsaa.
Se tapahtuu myös korean kanssa Persoonallisuus muuttuu kuten tantrumit ja huomiohäiriöt sekä petokset psykoosien yhteydessä. Älyllisen suorituskyvyn lasku johtaa progressiiviseen dementia (Hankittu henkinen vamma, katso siellä). Huntingtonin tauti on kohtalokas 15-20 vuoden kuluessa diagnoosista, usein seurauksena potilaan huonosta yleisestä tilasta johtuvista toissijaisista sairauksista.
Mitkä ovat ensimmäiset merkit?
Ensimmäiset Huntingtonin taudin merkit havaitaan yleensä 30–40-vuotiaina. Psykologiset valitukset edeltävät usein taudille ominaisia liikuntahäiriöitä vuosien varrella. Tyypillisiä psykologisia poikkeavuuksia ovat masennus ja vähentynyt ajotapa. Joskus alkavat kognitiiviset puutteet osoittavat keskittymis- ja muistihäiriöitä. Nämä oireet voidaan helposti sekoittaa masennukseen varhaisessa vaiheessa. Se, että tauti johtaa usein impulsiiviseen ja vahingolliseen käyttäytymiseen muita ihmisiä kohtaan, on stressaavaa myös sukulaisille.
Potilaat voivat saada osittain visuaalista tietoa, esim. Kasvoilmaisut eivät enää prosessoidu oikein, eivätkä siten enää reagoi asianmukaisesti muiden tunteisiin. Liikehäiriöille on alun perin ominaista hyperkinesia (Kreikan hyper - noin, kinesis - liike). Tämä tarkoittaa lisääntyneitä ei-toivottuja liikkeitä. Lihasääni - lihaksen jännityksen tila - vähenee. Potilaiden mielestä tämä oman kehon hallinnan puute on erittäin stressaavaa. Toisinaan, etenkin varhaisessa vaiheessa Itsemurhayritykset.
Kuinka tauti etenee?
Huntingtonin tauti on yksi kroonisesti etenevä neurodegeneratiivinen Sairaus. Tämä tarkoittaa, että se etenee yleensä hitaasti, mutta jatkuvasti, tuhoaa hermoja ja johtaa lopulta potilaan kuolemaan. Psykologisten poikkeavuuksien lisäksi liikuntahäiriöt ovat ominaisia sairaudelle. Varhaisissa vaiheissa tapahtuu yleensä enemmän ei-toivottuja liikkeitä (hyperkinesia) päällä. Ajan myötä yksi kehittyy hypokinesia. Kirjaimellisesti käännettynä tämä tarkoittaa "vähemmän liikuntaa", tarkoitetaan liikunnan puutetta, kuten on tyypillistä myös Parkinsonin oireyhtymässä. Taudin edetessä potilas tarvitsee yhä enemmän hoitoa. Progressiivinen dementia johtaa aluksi kielen köyhyyteen ja epäjärjestykseen. Ruoan saanti vaikeutuu yleensä nielemishäiriöillä, ja potilaat laihtuvat. Keskimäärin potilaat kuolevat 10–15 vuotta taudin puhkeamisen jälkeen. Jos taudin puhkeaminen tapahtuu myöhään, taudin kulku viivästyy usein jonkin verran.
Onko parannuskeinoa?
Huntingtonin tautia ei tällä hetkellä voida parantaa. Vuodesta 1993 tiedämme, että taudin syy on viallinen geeni Kromosomi 4. Valitettavasti geneettistä vikaa tai sen seurauksia ei tällä hetkellä voida hoitaa millään tavalla. Siksi et voi pysäyttää taudin kulkua tällä hetkellä. Tietenkin tutkitaan intensiivisesti uusia terapeuttisia lähestymistapoja. Taudin geneettinen perusta on nyt hyvin tiedossa.Siksi asianomaiset ja heidän sukulaiset voivat vain toivoa, että tutkimus tekee jossain vaiheessa merkittävän läpimurron.
Mitkä lääkkeet auttavat?
Huntingtonin tauti johtuu geenimutaatiosta. Valitettavasti tällä hetkellä ei ole lääkkeitä, jotka hoitavat tätä syytä tai parantavat tautia. Eri oireita voidaan yrittää hoitaa lääkkeillä. Neuroleptikoja käytetään usein klassisia liikuntahäiriöitä vastaan. Masennuslääkkeet auttavat masentuneissa mielialoissa. Viime kädessä nämä lääkkeet eivät voi estää taudin etenemistä. Yrität vain hallita oireita hiukan paremmin lääkityksen avulla.
Miltä loppuvaihe näyttää?
Yleensä se on loppuvaihe 10–15 vuotta saavutettu sairauden puhkeamisen jälkeen. Potilaat ovat sängyssä ja tarvitsevat hoitoa ympäri vuorokauden. Nielemishäiriön takia, joka kehittyy sairauden edetessä, monet ovat hyvin laihtuneet (lääketieteelliset: kakektinen). Elintarvikkeiden nielemisen yhteydessä on myös pysyvä riski hengenvaarallisesta keuhkokuumeesta (Aspiraatiokeuhkokuume) on tulossa. Jos potilas ei voi enää niellä, on harkittava keinotekoista ravitsemusta. Psykologiset poikkeavuudet lisääntyvät myös taudin edetessä. Lopulta dementia on edennyt, potilaat menettävät kykynsä kommunikoida ja häiriintyvät.
Differentiaaliset diagnoosit
Samanlaisia oireita, jotka koostuvat liikuntahäiriöistä ja henkisestä heikkenemisestä, voi esiintyä Creutzfeld-Jakobin tauti, taudin myöhemmissä vaiheissa Kuppa ja tulehduksen Aivot esiintyä.
Mikä aiheuttaa Huntingtonin taudin?
Huntingtonin tauti on geneettinen sairaus. Syy on geneettinen vika. Proteiini (proteiini), joka aiheuttaa taudin, kutsutaan Huntiniksi. Sitä koodaava geeni on Kromosomi 4. Huntingiiniproteiinin mutaatio aiheuttaa erityisten hermosolujen kuoleman tietyillä aivoalueilla. Tämä on hitaasti etenevä prosessi, minkä vuoksi tauti on yksi ns. neurodegeneratiiviset sairaudet. Monia sairauteen liittyviä patologisia prosesseja ei ole vielä tutkittu täysin. Huntingtonin taudin tiedetään kuitenkin olevan yksi Trinukleotiditauti toimii. Terveillä ihmisillä tietty DNA-yhdistelmä toistuu DNA: ssa jopa 20 kertaa. Huntingtonin tautia sairastavilla potilailla tämä yhdistelmä toistetaan paljon useammin, välillä 60–250 kertaa. Seurauksena on, että geeniä ei voida enää lukea oikein ja Huntinin-proteiini on koottu väärin. Mitä enemmän tämä toisto tapahtuu, sitä aikaisemmin henkilö kokee oireita. Mitä enemmän toistoja voidaan havaita potilaalla, sitä vaikeampi sairaus on.
Diagnoosi:
Kokoelma sairaushistoriaa ja kysymyksiä Huntingtonin taudin esiintymisestä perheessä. Fyysinen tutkimus keskittyen hermostoon.
Aivotoiminnan mittaus (EEG), mahdollisesti pään tomografia (leikkausröntgen). Koska geneettisen testin taustalla olevat muutokset tunnetaan, geneettinen testi voi luotettavasti diagnosoida ja jopa ennustaa Huntingtonin taudin. Tällainen ennustava (ennustava) diagnoosi on kuitenkin vain erittäin harvoin hyödyllinen, koska tauti ei tällä hetkellä ole parannettavissa ja siksi sillä ei olisi terapeuttisia seurauksia.
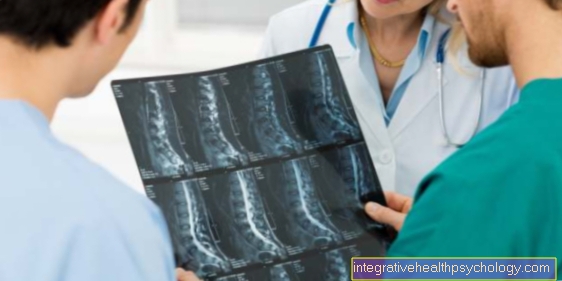
Aivojen MR
Jos epäillään Huntingtonin tautia, on järkevää ottaa poikkileikkauskuva aivoista. Tauti on neurodegeneratiiviset Sairaus, jossa hermosolut kuolevat prosessin aikana tietyillä aivoalueilla. Tämä näkyy myös MRI-kuvissa. Kudosten surkastuminen on erityisen ilmeistä alueella, joka vastaa vapaaehtoisesta liikkeestä. Näin Sivukammio (= aivovesillä täytetyt ontelot) laajentunut kuvantamisessa. Tämä on suhteellisen klassinen havainto Huntingtonin taudille. Lopullinen diagnoosivarmuus saadaan geneettisellä testillä (katso kohta tästä).
Kuinka Huntingtonin tauti periytyy?
Huntingtonin tauti on yksi autosomaalinen hallitseva perinnöllinen sairaus. Jos geeni periytyy hallitsevasti, se tarkoittaa, että se on jo viallinen alleeli toisessa kahdesta kromosomit johtaa ominaiseen ilmaisuun. Termi autosomaalinen johdetaan autosomeista. Kaikkia kromosomeja, jotka eivät osallistu sukupuolen määrittämiseen, kutsutaan autosomeiksi. Tämä tarkoittaa, että perintö on riippumaton sukupuolesta. Joten voit periä viallisen geenin molemmilta vanhemmilta. Siksi miehet ja naiset vaikuttavat yhtäläisesti. Huntingtonin taudin tapauksessa viallinen geeni on päällä Kromosomi 4. Vaikka perintö ei ole sukupuolesta riippumatta, on osoitettu, että tauti alkaa aikaisemmin ja että sen eteneminen on dramaattisempaa, jos viallinen geeni periytyy isältä. Äidin perinnöissä on kuitenkin todennäköisempää, että sairaus alkaa myöhemmin.
Geneettinen testi
Huntingtonin taudista vastuussa oleva mutatoitu geeni paljastetaan Kromosomi 4. Se löydettiin vuonna 1993. Sen jälkeen on ollut saatavilla geneettinen testi. Joten jos potilaalla epäillään olevan Huntingtonin tauti, verinäyte voidaan tutkia sen selvittämiseksi, onko potilaan DNA: ssa tämä mutaatio. Tämä turvaa diagnoosin. Terveillä ihmisillä, joilla on rakkaansa Huntingtonin taudissa, voidaan myös veri testata mutaation suhteen. Huntingtonin tauti on perinnöllinen sairaus. Tällä on usein kauaskantoisia vaikutuksia kärsivien ihmisten elämään, minkä vuoksi on olemassa erityiset ohjeet terveiden ihmisten geenitesteille. Esimerkiksi. alaikäisiä ei testata; geneettisiä testejä ei saa suorittaa kolmansien osapuolten (vanhemmat, kumppanit, ...) pyynnöstä. Havaitsemalla geenimutaation terveillä ihmisillä, diagnoosia ei ole heti, mutta jos saavutetaan tietty määrä DNA: n tietyn sekvenssin toistoja, sairastuneelle henkilölle todennäköisesti kehittyy Huntingtonin tauti sairauden aikana.
Therapy:
Huntingtonin taudin syyn hoitaminen ei ole tällä hetkellä mahdollista. Liialliset liikuntahäiriöt voidaan estää lääkityksellä. Tietyissä olosuhteissa psykoterapian seuraaminen tai liittyminen omatoimiseen ryhmään voi auttaa potilasta käsittelemään tietoa sairaudesta.
dementia
Klassisten liikuntahäiriöiden lisäksi Huntingtonin tauti johtaa myös psykologisiin muutoksiin. Nämä ovat Vaikuttaa (= Tunnelma kiihtyy masennukseen), mutta myös kognitiiviset rajoitukset. Ne ilmestyvät usein varhaisessa vaiheessa muistin häiriöinä. Potilaan älylliset kyvyt heikentyvät alussa vain hieman; ulkopuoliset eivät usein huomata tätä. Taudin edetessä kognitiivisten kykyjen menetykset kasvavat aina dementiaan saakka. Puheen köyhtyminen tapahtuu ja potilaat ovat usein täysin häiriintyneitä.


.jpg)



.jpg)